Small-angle X-ray scattering from solutions of biological macromolecules, also known as BioSAXS, is a structural biology technique of rapidly increasing popularity. Other than with single crystal X-ray diffraction, SAXS allows to study proteins in their native state, dissolved in a suitable buffer, and crystallization is not required.Other than with single crystal X-ray diffraction, SAXS allows to study proteins in their native state, dissolved in a suitable buffer, and crystallization is not required. BioSAXS has proven as a valuable tool to analyze the overall size, compactness and aggregation behavior of protein molecules, and it can even be used to determine the 3D envelope structure at a resolution of 1-2 nm.
SAXS measurements were performed on a dilute solution of glucose isomerase. By using ScatterX78 experimental data could be acquired with good sensitivity and within short measurement times. The EasySAXS data analysis software was used to deduce information about the size, compactness, overall shape and aggregation behavior of the protein molecules. All results were found to be consistent and in good agreement with what has been reported in the scientific literature.
Small-angle X-ray scattering from solutions of biological macromolecules, also known as BioSAXS, is a structural biology technique of rapidly increasing popularity. Other than with single crystal X-ray diffraction, SAXS allows to study proteins in their native state, dissolved in a suitable buffer, and crystallization is not required. BioSAXS has proven as a valuable tool to analyze the overall size, compactness and aggregation behavior of protein molecules, and it can even be used to determine the 3D envelope structure at a resolution of 1-2 nm. Glucose isomerase is a protein that is often used as a benchmark for the validation of BioSAXS data quality and of data analysis. Its dimensions and structure are well documented in the scientific literature [1-3]. In its native state the protein exists as a tetramer with a molecular weight of 173 kDa. Glucose isomerase was used to validate the performance of ScatterX78 in this type of challenging application.
Glucose isomerase from Streptomyces rubiginosus (Hampton Research) was dialyzed against a buffer made of 100 mM Tris and 1 mM MgCl2 having a pH of 8.0.
The final protein concentration was 1.1 wt.%. The protein solution was loaded in a disposable quartz capillary with a diameter of 1 mm for which a sample volume of 50 µl is required. For the background measurement the same capillary filled with the pure buffer was used. SAXS data were acquired on the Empyrean platform configured with ScatterX78 in combination with a focusing X-ray mirror and the PIXcel3D detector. Measurements were performed at room temperature using a 60 min scan. For comparison a quick 10 min measurement with the detector in static mode was also done. The EasySAXS software was used for data reduction and analysis.
In spite of the relatively low scattering intensity it was possible to observe a distinct excess scattering signal from the dissolved protein molecules. Meaningful data could be readily obtained at scattering vectors q ranging from as low as 0.06 nm-1 (or 0.08 deg 2θ) up to 3.6 nm-1.
The Guinier plot (ln(I) vs. q2) of the experimental data acquired at the smallest angles shows that the data points fall on a straight line. The radius of gyration Rg, which may be seen as an overall size parameter of the protein, can be calculated from the slope to 3.31 nm. The value for Rg from the 10 min measurement (3.33 nm) is already in good agreement with the 60 min measurement.
Furthermore, there are no signs of unspecific aggregation of the protein molecules, which would be evidenced by an increase of the intensity at the smallest angles.

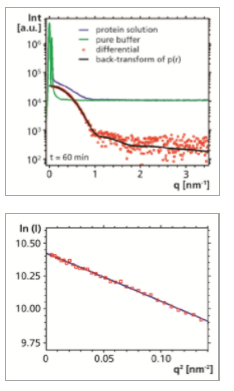
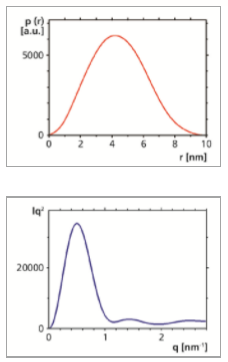
By using an indirect Fourier transformation procedure the pair distance distribution function p(r) was determined. It was found to be well-behaved, with an overall symmetric shape, which indicates that also the protein has an overall symmetric shape. It approaches zero at a distance r of approximately 9.7 nm. This value may be interpreted as the maximum dimension Dmax of the protein molecule. It is in good agreement with the value given in the literature [2]. The calculation of Rg based on the second moment of p(r) yielded a result that is consistent with Rg determined from Guinier analysis. Furthermore, the values for Rg and Dmax determined from the 10 min and 60 min measurements are in good agreement (see Table). Also the backtransformation of the p(r) function is a good approximation of the experimental data.
By displaying the (non-smeared) SAXS data in a Kratky plot (Iq2 vs. q), qualitative information about the compactness of the protein can be deduced. Here the non-smeared data obtained from the back-transformation of the p(r) function have been used. A bell-shaped curve is observed, indicating that the protein has a compact, folded structure, rather than an unfolded (random coil) structure.
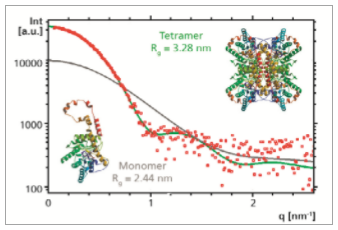
Based on the published structure of glucose isomerase determined by single crystal X-ray diffraction [3] it is possible to simulate the corresponding solution scattering (SAXS) data. This was done both for the monomeric unit and for the tetramer, by using the software CRYSOL [4]. Comparison with the experimental SAXS data shows that there is a good agreement with the simulation that was done for the tetramer. Also Rg determined from the experimental data (3.31 nm) agrees very well with the Rg obtained from this simulation (3.28 nm). From this it can be confirmed that in solution the protein forms a tetramer rather than a monomer, and that its structure in solution is very similar to its crystal structure.
ScatterX78 on the Empyrean platform delivers high-quality SAXS data within short measurement times, even from dilute protein solutions. The performance of the experimental SAXS setup and of the EasySAXS data analysis software was successfully validated, yielding consistent results that agree with published data.