A review of the methods available for measuring the key characteristics of polymers focusing on the benefits and value of gel permeation/size exclusion chromatography (GPC/SEC). Much of the paper talks exclusively about polymers, however many of the principles discussed are equally applicable to proteins or protein conjugate materials.
Polymers are ubiquitous throughout industry, in the form of naturally occurring materials, such as cellulose and starch, and synthetic commodities - polystyrene, polyethylene and nylon, for example. As polymer technologies advance, the strength, stability, chemical resistance, and performance modifying properties of these industrially vital materials are meeting increasingly diverse applications. Accurate and precise polymer characterization is essential from R&D through to QC, to ensure that advanced materials meet the exacting performance targets expected of them.
The functionality of polymers is defined by their molecular weight (MW), MW distribution, molecular size and structure, the degree of chain branching or cross linking. Effective and efficient methods for measuring these properties are therefore an essential part of the polymer developer’s analytical toolkit.
This whitepaper reviews the methods available for measuring the key characteristics of polymers focusing on the benefits and value of gel permeation / size exclusion chromatography (GPC/SEC). Much of the paper talks exclusively about polymers, however many of the principles discussed are equally applicable to proteins or protein conjugate materials.
A good starting point when looking at methods for polymer characterization is to understand what a polymer is and how this influences the techniques that are suitable for measuring polymer properties, most especially MW.
The term polymer derives from the Greek words ‘poly’ and ‘meros’ meaning many parts and it highlights a defining feature of polymeric materials which is their chain like structure. This structure is formed through the development of chemical links between multiple repeating units or monomers. For example, polymerizing styrene (a monomer), under appropriate reaction conditions, produces the polymer polystyrene (see figure 1).
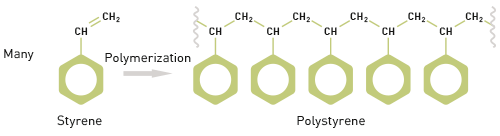
|
The molecular weight of a polymer therefore relates to that of the monomer and to the number of monomer units in the polymer molecule. Styrene has a molecular weight of 104 Da and so polystyrene has a molecular weight of 104 multiplied by n, the number of styrene molecules in the polymer chain. However, polymer samples rarely contain chains that are all of identical length, rather there is a distribution of chain lengths.
This distributive feature of polymers complicates any discussion of MW, necessitating the use of averaged figures, and the associated measurement of distribution. Both the shape of the distribution and average MW affect the properties exhibited by a polymer. Determining MW therefore requires measurement of both the MW of individual chains and the number of chains of any particular weight.
Generally speaking polymer MW exhibits a distribution as shown in figure 2. Using statistical mathematics it is possible to define three different moments for this distribution each of which can be considered as an average MW. The two most commonly used terms are Mn and Mw, the number and weight averaged MW respectively. Mn is the midpoint of the distribution in terms of the number of molecules. Mw, in contrast, describes the midpoint of the distribution on the basis of weight. Mz, the third moment, is a figure more weighted towards higher MWs. The ratio of Mw to Mn is referred to as the polydispersity and is routinely used to describe the width of the distribution.
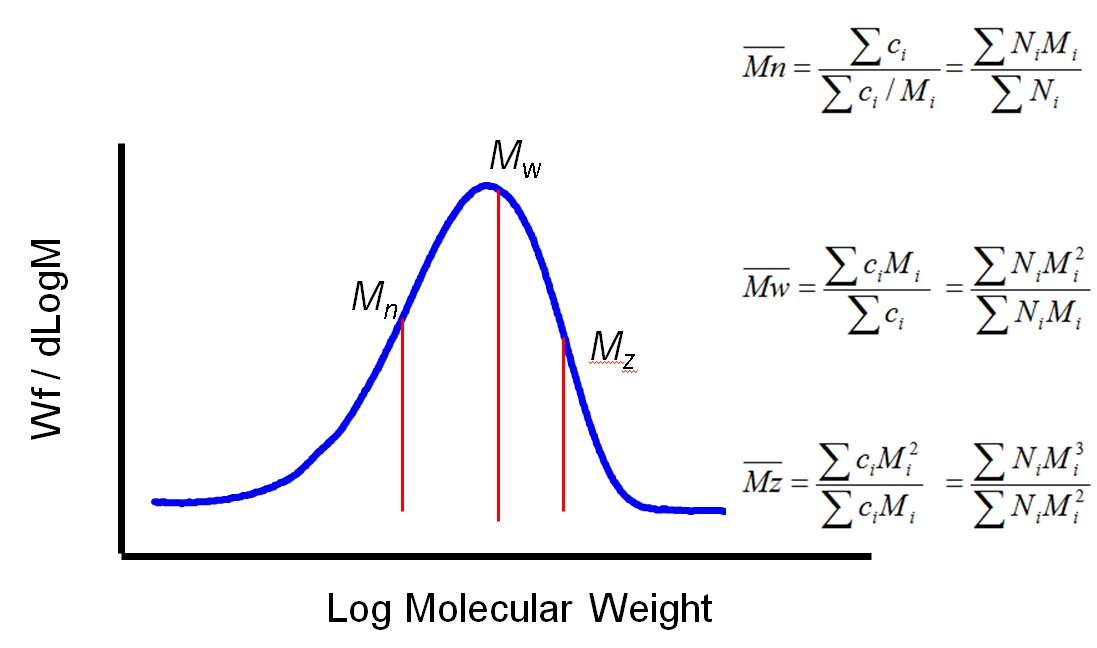
|
When it comes to measuring MW, the term ‘absolute’ MW is often used. The preceding analysis makes clear that any absolute measurement of MW must also involve the absolute measurement of concentration or number of molecules. This is an important point to consider when assessing alternative techniques for MW measurement.
There are many different techniques that can be used to measure MW including: end group analysis, membrane osmometry, viscometry and light scattering. However light scattering, more specifically Static Light Scattering (SLS), is increasingly the standard approach. For a fuller description of SLS please refer to the whitepaper ‘Static light scattering for GPC-SEC explained’, see reading list below, however a brief description of the principles is provided here.
When a macromolecule is irradiated by an incident beam of light photons are absorbed and then re-emitted or scattered in all directions. The intensity of the scattered light is proportional to the MW of the polymer, a relationship described by the Rayleigh equation.
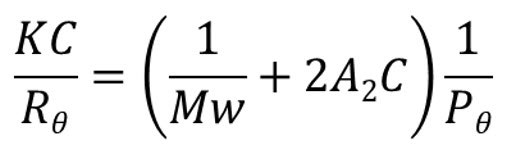
|
The Rayleigh equation becomes relatively simple to apply when the term Pɵ, which describes the angular dependence of the scatted light, tends to 1. This occurs when ɵ becomes zero i.e. the scattered light is measured at an angle of zero degrees to the incident beam. Unfortunately, measuring at an angle of 0o is not practical as the incident beam intensity dwarves the signal intensity from the scattered light, preventing its determination.
Small molecules, less than around 10 – 15 nm radius, scatter light isotropically (Pɵ=1), so intensity measurements are independent of the angle at which the measurement is made. However, larger molecules scatter anisotropically (Pɵ<1), a phenomenon that must be accounted for to access accurate MW for this class of molecules using the Rayleigh equation. Three different approaches to SLS have been developed to address this issue: right angle light scattering (RALS); low angle light scattering (LALS); and multi-angle light scattering (MALS):
With RALS, scattering intensity is measured at 90o to the incident beam. This gives an excellent signal to noise ratio. However a RALS detector takes no account of anisotropic scattering, it simply assumes that the scattering intensity at 0o is the same as at 90o. This approach is good for small molecules but can give unpredictably inaccurate results for anisotropic scatterers.
LALS is based on the measurement of scattered light at an angle very close to 0o to eliminate the error associated with anisotropic scattering. This works for all molecules but the signal to noise ratio becomes challenging for smaller molecules. Combining RALS/LALS technology can therefore work well.
The approach adopted with MALS is to measure at multiple angles and then extrapolate to find a value for scattering intensity at 0o. MALS works for all sizes of molecules but the technique is more complex than either RALS or LALS and the associated detectors are more expensive.
All three light scattering techniques can be used in batch mode, but are more commonly applied in flow mode, with the light scattering detector forming part of a gel permeation/size exclusion chromatography (GPC/SEC) detector array.
The development of GPC/SEC in the mid 1960s1 brought a powerful new tool to polymer scientists that for the first time enabled detailed study of each part of a polymer distribution. GPC/SEC begins with size fractionation of a sample, followed by detection of each fraction of the sample as it elutes from the separation column (see figure 3). The only drawback with GPC/SEC is that separation is based on the size of the polymer molecules, not on their MW.
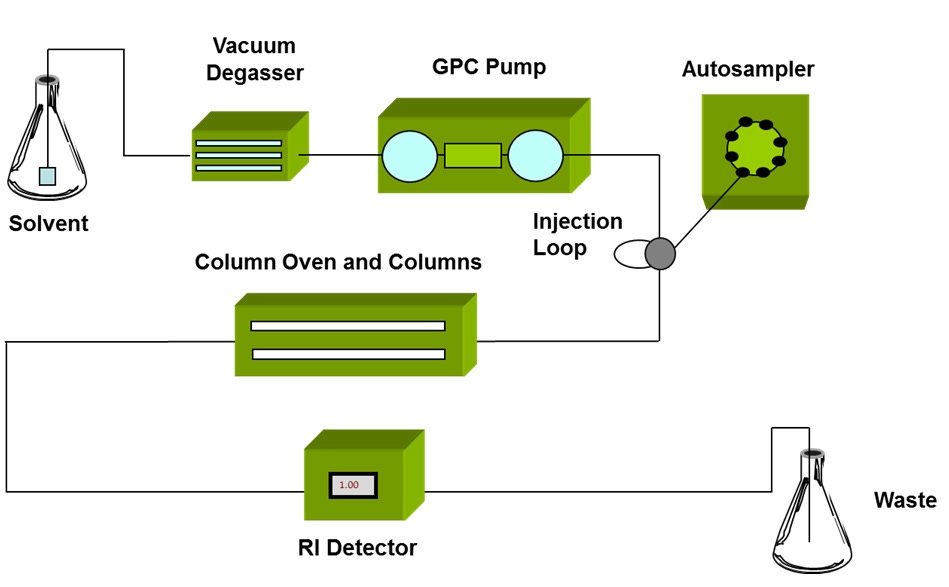
|
This limitation can be tackled in a number of different ways, using conventional calibration, universal calibration, or by the addition of a light scattering detector to directly measure MW. In GPC/SEC, the term ‘absolute’ MW is, as a result, now used to differentiate a method that directly measures MW from one that infers it, typically from calibration techniques that are reliant on the use of a set of relevant standards.
Light scattering detectors are a useful and convenient tool for measuring MW within a GPC/SEC system set-up. They do not in the strictest sense measure ‘absolute’ MW, since all of the light scattering techniques involve instrument calibration. In addition, MALS detection is dependent on a model fit. However, SLS delivers by far the most accurate data available with existing technology and can be considered ‘absolute’ in the sense of direct measurement. When selecting the best light scattering for any given application it is crucial to closely consider the available options and the strengths and limitations of each light scattering approach. Further guidance on this topic is provided in the webinar ‘Polymer solution characterization – Part 1’ (see further information below for more details).
Molecular size, like MW, is a defining characteristic of most polymers. From a practical perspective the size of a polymer molecule in solution directly influences its rheological behavior which is often directly linked with formulation performance. However, size measurements are equally valued because of the understanding they provide about the conformational changes a polymer undergoes in a specific environment, the extent of solvation and solubility, and any branching in the polymer chain. This insight can be used to establish the structure-function relationships required for successful commercial application.
The relationship between molecular size and MW is not constant, different polymers exhibit different molecular densities in solution. The need for size data therefore calls for independent measurement. The size a polymer attains in solution is influenced by the solvent in which the polymer is being handled, its type, temperature and pH, and also by factors that affect the way in which polymer conformation or folding occurs. This behavior is a function of the polarity of the polymer and its structure, most especially any steric effects caused by bulky groups attached to the polymer chain.
Most polymer molecules are <100nm so the need for size information calls for techniques with the capability to measure nanoparticles. Three different methods are routinely applied: dynamic light scattering (DLS); MALS; and SLS in combination with intrinsic viscosity (IV) measurement.
In contrast to SLS, DLS measurements derive from the real-time fluctuations in scattered light, rather than the time-averaged data that are the focus with static techniques. For a fuller description of DLS please refer to ‘Dynamic Light Scattering: An Introduction in 30 Minutes’, see reading list below, however a brief description of the principles is provided here.
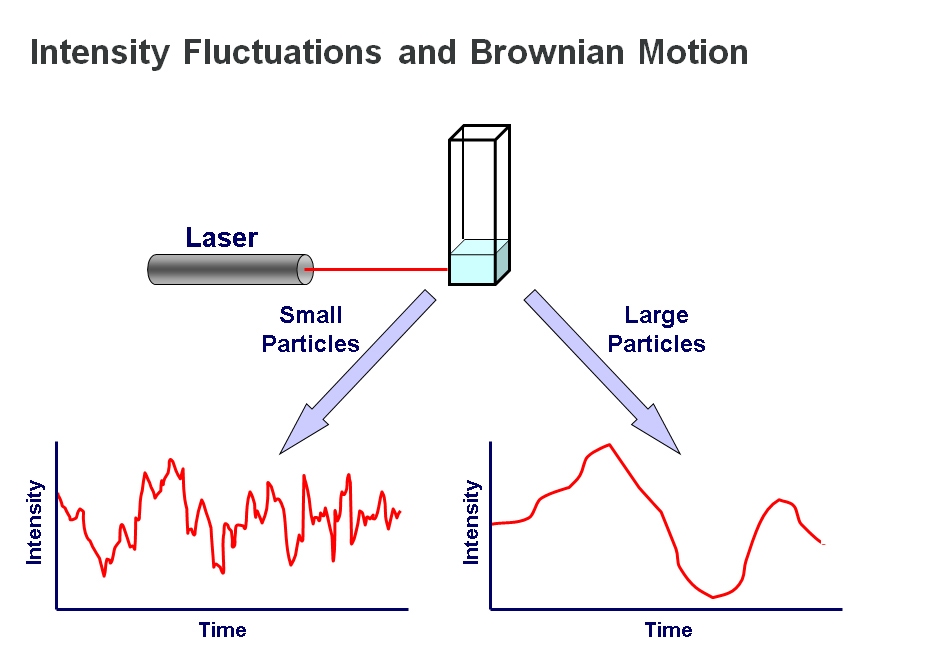
DLS is most often applied in batch mode. The sample is illuminated by a laser beam, causing light scattering which is detected by a sensitive photon counting module. By using a correlator, the detected fluctuations in light scattering intensity can be translated into a measure of diffusion speed, thereby providing a value for molecular or particle size.
The underlying principle of the technique is that small particles move relatively rapidly under Brownian motion while larger particles move more slowly. The relationship between size and speed of motion is well understood and described by the Stokes-Einstein equation2. The quicker movement of smaller particles is reflected in relatively rapid fluctuations in scattered light intensity while larger particles, conversely, are associated with fluctuations over a longer timescale. Measurements of scattered light intensity therefore enable the determination of particle or molecular size, more specifically hydrodynamic radius (Rh).
|
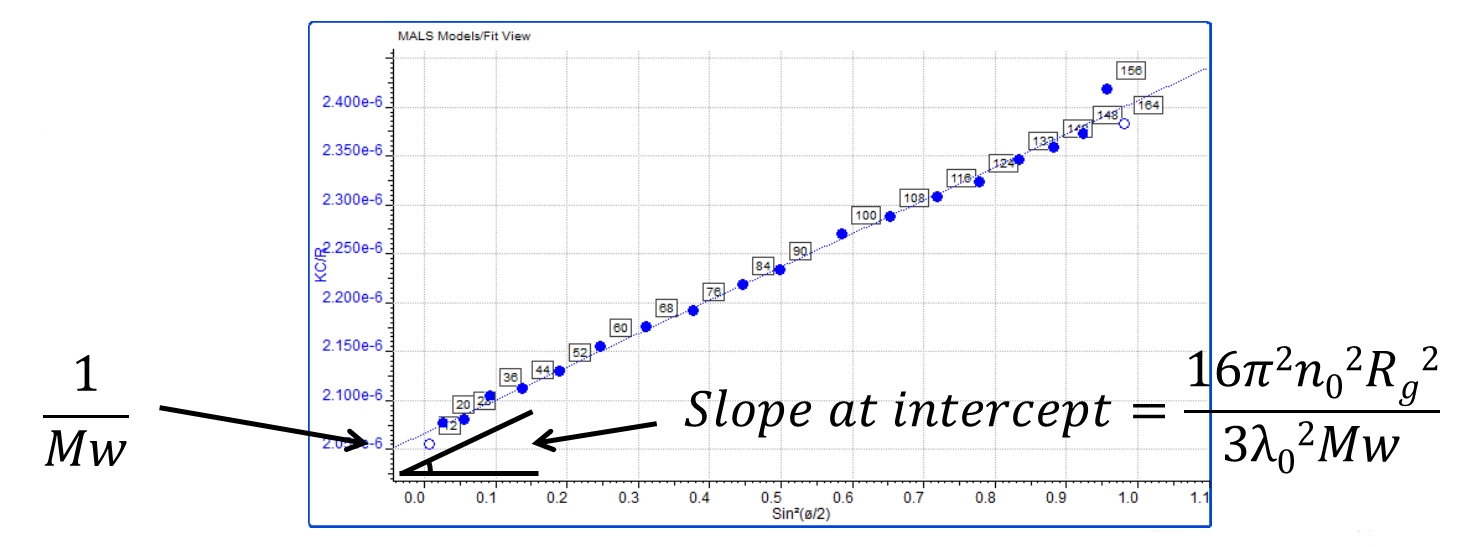
The measurement of molecular size by MALS exploits the fact that the anisotropic light scattering pattern produced by larger molecules, approximately 10 – 15 nm radius and above, is related to their size. This means that molecular size cannot be measured by MALS for smaller molecules. However for larger molecules it is possible to calculate a size parameter, radius of gyration (Rg). Rg is actually the average squared distance of any point in the polymer coil from its center of mass and it can be calculated from the initial slope of the Zimm plot where it intercepts the axis. This is produced by plotting functions of light scattering intensity against angle.
|
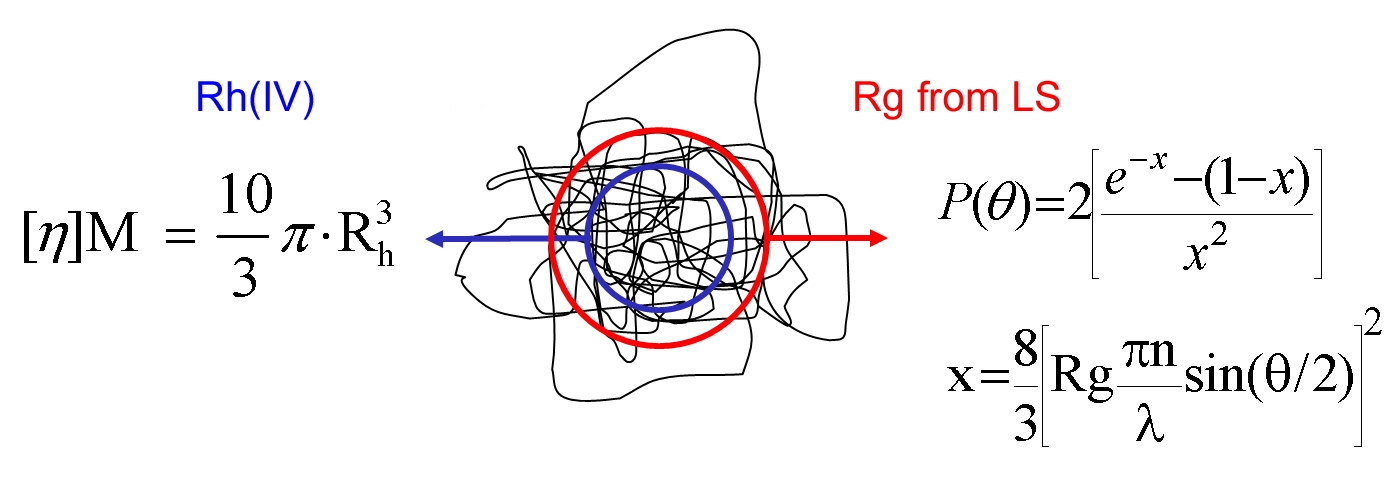
Although the relationship between the MW and molecular size of a polymer is not constant it can be measured. The parameter IV, which has units of dl/g, can be determined using viscometry measurements (see below) and directly relates molecular size to MW for any given polymer. In other words IV is an inverse measure of molecular density. Less tightly coiled polymers have a relatively high IV while those that densely entangle have a relatively low IV. Molecular size can therefore be calculated from MW measurements by SLS combined with IV.
This method generates values for hydrodynamic radius (Rh), however it is important to note that although this name suggests that the parameter is equivalent to that measured by DLS this is not technically true, because size is derived in a different way in each case. Rh is also different from Rg (see figure 6).
|
The three different methods outlined for molecular size measurement all deliver a different size parameter, Rh (DLS), Rg and Rh(IV). This raises the question of whether the size parameters are in close agreement and if not, why not?.
In reality, the magnitude of any numerical differences in these three size parameters depends on the type of molecule being measured, on its molecular density and shape. For example, measurements of Rh(DLS) and Rh(IV) made for BSA, a dense (low IV), globular molecule, show close agreement. In an experimental study of this material Rh was found to be 3.1 nm by DLS and 3.3 nm by the SLS + IV method. At around 3 nm the size of this molecule is too small to be measured by MALS so here only two techniques are applicable and they provide similar results.
In contrast, comparable analysis of a very different type of polymer, a complex polysaccharide with a non-globular, low density structure reveals significant differences between the different size metrics. In this experiment Rg could be measured and was found to be 36 nm. In contrast, Rh (IV) was found to be 24 nm and Rh ( DLS) 20 nm. Rg is quite differently defined from Rh and would therefore be expected to provide a different and indeed larger result (see figure 6). The differences in Rh on the other hand are arguably less expected but derive from the effect of polymer shape and density, differently impact each measurement technique.
One final point to note about molecular size is that it is, like MW, a distributed parameter. Using the outlined methods within a GPC/SEC system therefore enables the measurement of polymer size distribution in place of the ensemble or averaged data produced by a batch measurement.
The identified disconnect between the MW and molecular size of polymer stems from the structural characteristics that polymers can exhibit. Polymers can be linear or branched, and in addition can cross-link, where one part of the polymer chain chemically reacts with another. All of these features influence the behavior of a polymer during use. Quantifying and understanding these characteristics is therefore essential for development of the structure-function relationships that underpin successful polymer use.
Using GPC/SEC and the outlined measurement methods it is possible to measure MW and molecular size distributions for a polymer. The elucidation of structural characteristics relies on using these data in combination with measurements of IV, which as noted, is a measure of molecular density. The table below summarizes the key ways in which IV is altered by changes in structure, relationships which underpin the value in adding a viscometer to a GPC/SEC system to determine IV and thereby elucidate structure.
Structural or conformational change | Effect on density | Effect on IV |
|---|---|---|
a) Increase chain length (MW) of linear molecule. | Decreases | Increases according to Mark-Houwink equation |
b) Increase mass of chain segments, keeping chain length constant. | Increases | Decreases |
c) Increase stiffness of chain. | Decreases | Increases |
d) Add branches to chain, keeping MW constant. | Increases | Decreases |
e) Collapse chain into dense molecule. (natural protein or aggregate) | Increases greatly | Decreases greatly |
The viscosity of a polymer solution (ɳ) derives from the viscosity of the solvent (ɳ0) but includes a contribution from the polymer. This contribution depends on the concentration of polymer in solution. Two routinely used dimensionless viscosity parameters defined on the basis of the different between polymer solution and solvent viscosity are:
Both of these parameters are concentration dependent. IV in contrast is not and provides a more fundamental measure of the contribution that a polymer will make to polymer solution viscosity. It is a measure of the volume of solution occupied by a single molecule when it is not interacting with any other molecules (low concentration state) and is therefore simply dependent on solvent and temperature, both of which should be quoted alongside any measured values. IV is related to specific viscosity by the following equation:
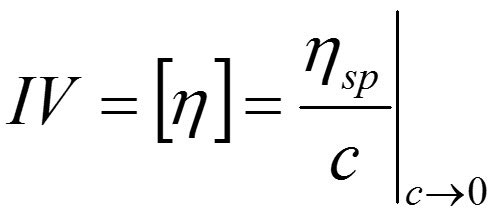
IV can be determine by batch techniques or in a continuous mode, within a GPC/SEC set-up, but in either case it is specific or relative viscosity that is actually measured. Batch methods are discussed in the presentation ‘Polymer solution characterization – part 3’ (see further information below), but the focus here is the measurement of IV using a four capillary viscometer of the type routinely applied in GPC/SEC set-ups.
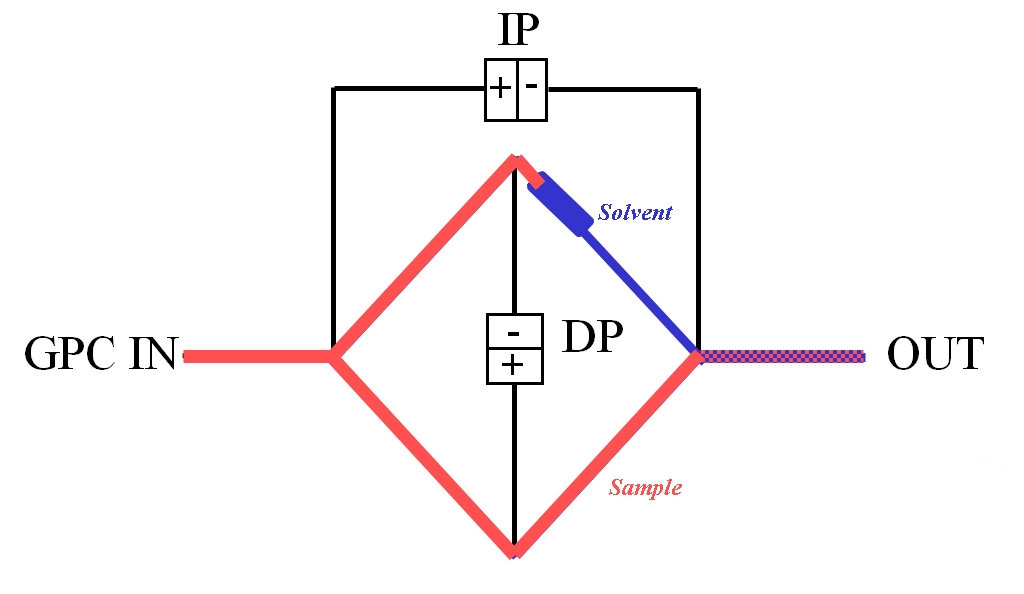
|
The viscometer shown in figure 7, a design specified for use within a GPC/SEC set-up, works in a similar way to a Wheatstone bridge in electronics, on the principle of balancing resistances. Differential pressure transducers measure both the pressure drop across the center of the bridge, DP, and from inlet to outlet, IP. Key design features are that the delay volume must be greater than the net elution volume of the separation column but with negligible flow resistance, and that all four capillaries present essentially equal resistance to flow. During baseline conditions the four capillaries and the delay volume are filled with pure solvent.
As the eluting sample flows into the viscometer it rapidly displaces solvent from all of the capillaries except for the one after the delay volume, which now acts as a reference. The readings of IP and DP enable calculation of the pressure drop resulting from flow of the solution relative to flow of the pure solvent in the reference. This is converted into viscosity data via Poiseuille’s Law, which correlates the pressure drop across a tube with its physical dimensions, the flow rate of fluid through it, and viscosity. Such calculations yield specific viscosity from which IV is directly calculated.
While differences in IV can be used to infer information about structure using the general correlations outlined in table 1, more detailed information about structural characteristics can be determined from a Mark-Houwink (M-H) plot, a log-log plot of IV against MW, both of which we now fully understand how to measure.
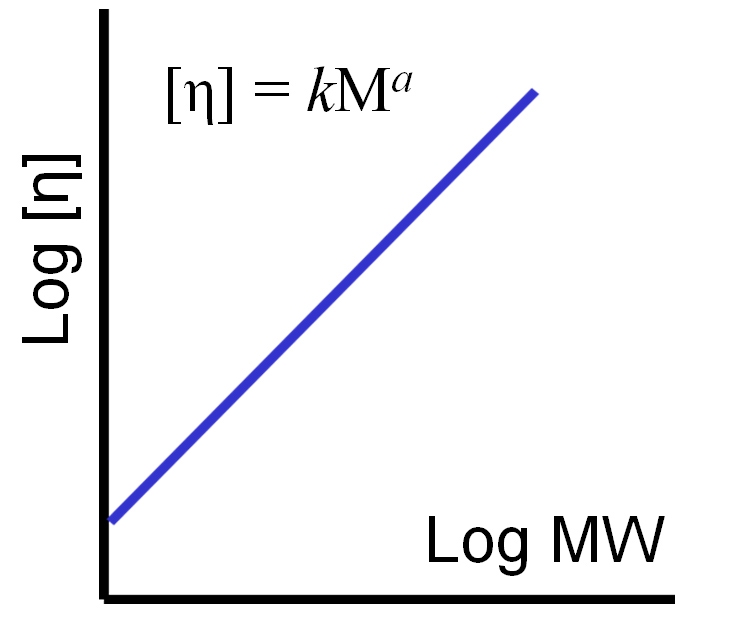
|
From a M-H plot it is possible to determine the M-H parameters, k and a as shown in figure 8. These parameters are often referred to as M-H ‘constants’ but in fact, k and a are not constant, but rather vary considerably depending on the structural characteristics of the polymer. An assessment of these parameters by examining the complete M-H plot can therefore be extremely helpful in investigating the structural features of a polymer.
For example, figure 9a shows an M-H plot for a relatively high MW, standard linear polystyrene in THF. This was generated in a GPC/SEC experiment by precisely measuring the MW (light scattering) and IV (viscometer) of the eluting sample. The result is a highly accurate, straight line M-H plot, from which values of k and a can be easily determined. The straight line indicates the polymer structure in solution is unchanging across all the molecular weights it contains.
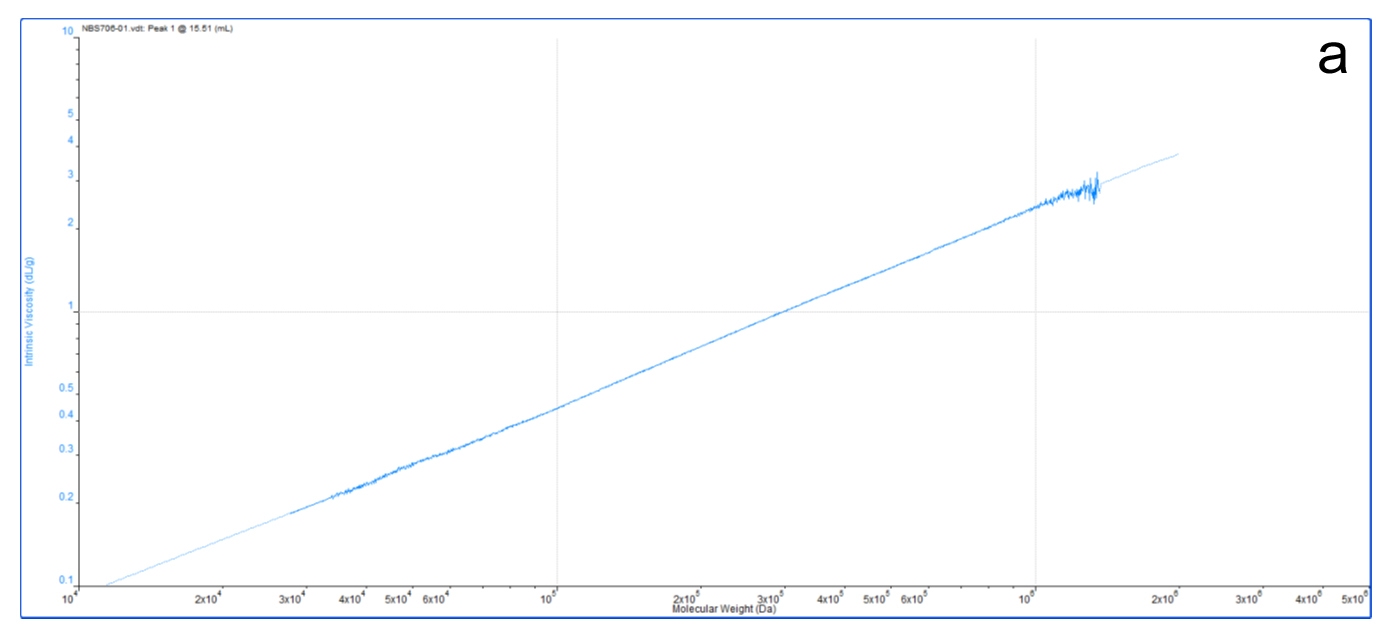
|
|
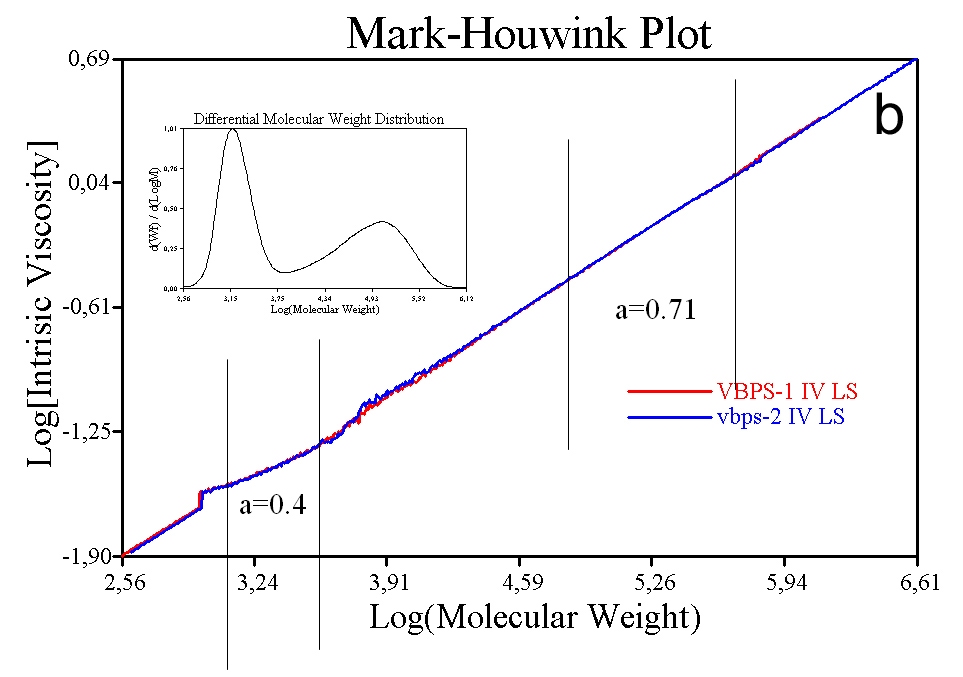
Figure 9b shows the same data for a sample containing some low MW polystyrene in addition to the original material. Even though the added material is the same chemical (polystyrene) and structural (linear) composition a difference is observed in the M-H plot and the associated values of k and a. The shallower slope of the plot in the low MW region is attributable to differences in the way in which the low MW polystyrene is solvated. These results highlight how the M-H ‘constants’ can change, even for a specific polymer, and the associated need to measure these constants rather than relying on published data.
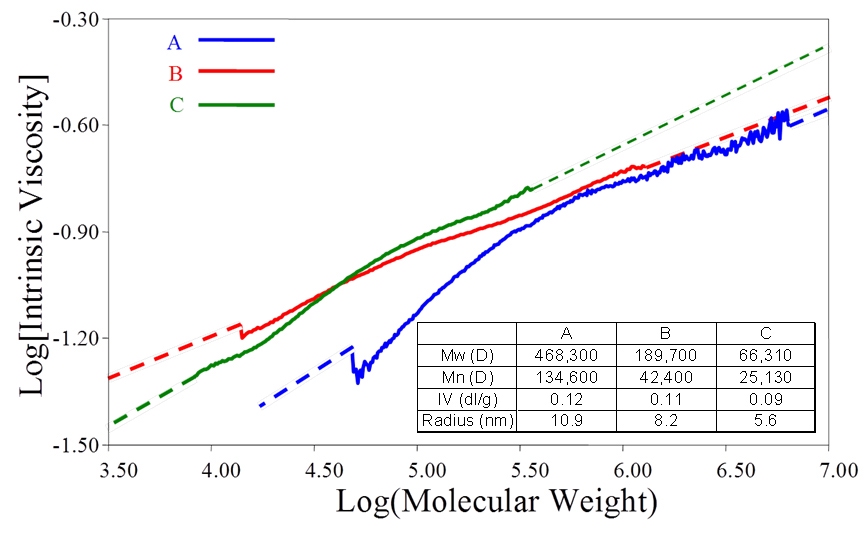
M-H plots are extremely useful for comparing the similarity or otherwise of samples, in terms of, for example, the extent of branching. Figure 10 shows plots for three batches of Maltodextrin, the properties of which are shown in the inset table. The bulk IV values of these samples is very similar, an important point since if bulk viscosity was used as the QC tool these samples would appear to be identical. These are actually food additives and would give very different performance in a formulation, despite their similar IV values. The MW results highlight marked differences between the three samples, which are graphically illustrated and elucidated via the associated M-H plot. Sample A has a lower IV at lower MW but also contains much higher MW material than the other two samples. This indicates that sample A is much more highly branched than the other two samples.
|
Although assessments of the degree of branching in a polymer often extend only as far as a comparison of M-H plots it is possible to numerically quantify the extent of branching in a polymer using the parameters already discussed. Work carried out by Zimm and Stockmayer3 provides a relationship for branching defined in terms of Rg. This can be manipulated to enable the calculation of branching ratio from measurements of IV, though a shape factor is required to successfully apply this approach. However, generally IV data can be measured with far greater accuracy and range than Rg so it is the most practical method available. For a fuller explanation of branching calculations and to see a worked example please refer to the webinar ‘Polymer solution characterization – Part 3’.
The comprehensive characterization of polymers underpins their successful application in a growing number of commercial applications. Key properties include MW, MW distribution, molecular size and molecular size distribution, and structural characteristics such as the extent of branching. The methods outlined here show how all of these properties can be measured with efficiency and accuracy, and highlight the central role of GPC/SEC for polymer development and QC.
References:
1 J.C. Moore ‘Gel permeation chromatography – A new method for molecular weight distribution of high polymers’ J. Polym. Sci., Part A-2, 835, (1964)
2. Einstein, A. ‘Investigations on the theory of the Brownian movement’, Fȕrth, R., editor, Cowper, A.D., translator. Methuen, London, 1926, p124
3. ‘The Dimensions of Chain Molecules Containing Branches and Rings’ J. Chem. Phys. 17, 1301 (1949)
Further information
All the topics discussed here are covered in greater detail by the webinar series ‘Polymer solution characterization’ Parts 1 to 3. These are freely available for viewing at: www.malvern.com
For more information of the different approaches to SLS read ‘Static light scattering for GPC-SEC explained’ http://www.malvern.com/slsexplained
For a fuller description of DLS please refer to ‘Dynamic Light Scattering: An Introduction in 30 Minutes’ http://www.malvern.com/en/support/resource-center/technical-notes/TN101104DynamicLightScatteringIntroduction.aspx