See how to use ITC to modulate Protein Protein interactions in the drug discovery process
Protein/Protein interactions define a large fraction of the validated targets for drug development including cancer, inflammation, diabetes, osteoporosis, infection and autoimmune diseases. Until now Protein/Protein interactions have been disrupted or replaced by other proteins. For example, antibodies or chimeric constructs are used to inhibit ligand/receptor interactions, and recombinant proteins are used as substitutes for deficient or insufficient natural proteins. The use of small molecules as antagonists or agonists of protein/protein interactions is still in its early days. The successful targeting of protein/protein interactions requires a precise characterization of the protein partners, their interaction and the allosteric consequences of that interaction. As illustrated in this chapter, Microcalorimetry is ideally suited to perform this characterization.
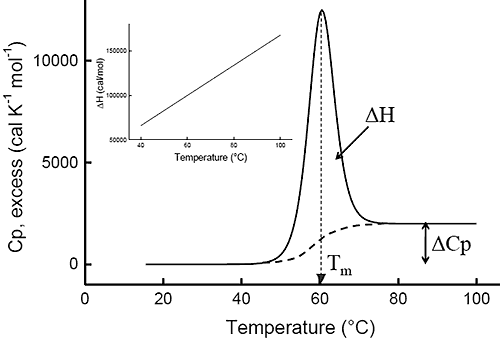
Ever since the pioneering work of Privalov in the seventies,1-4differential scanning calorimetry (DSC) has been used to determine the magnitude of the forces that stabilize the native structure of proteins. After proper data processing, the quantity measured in DSC is the heat capacity of a protein as a function of temperature. Figure 1 below shows a typical scan of a monomeric globular protein that undergoes a two-state transition (more complex transitions like multistate or irreversible denaturation can also be accurately treated).5-8 The disruption of interactions in the protein gives rise to an endothermic denaturation peak with its maximum value centered at the transition temperature, Tm, which often is used as a measure of protein stability. The graph shows the typical sigmoidal shift in baseline to a higher heat capacity at the completion of the denaturation. The enthalpy, ΔH, associated with the process corresponds to the area under the curve after subtraction of the sigmoidal baseline. The unfavorable enthalpy of transition is opposed by the large favorable entropy change, ΔS, associated with the increase in degrees of freedom gained when the restricted amino acids become unfolded. This conformational entropy increase is large enough to also overcome the unfavorable solvation entropy associated with the hydration of the protein core upon unfolding.
Furthermore, when the hydrophobic residues become exposed, water molecules form a highly structured hydration layer with a high heat capacity. The sigmoidal baseline change for the denaturation corresponds to this large and positive change in heat capacity, ΔCp. As a consequence of the large value for the change in heat capacity, both the enthalpy and the entropy changes for protein denaturation depend strongly on temperature. The inset in Figure 1 shows the relation between enthalpy and temperature.
|
The stability of the native state of a protein is dictated by the magnitude of its Gibbs energy (ΔG) which is given by the standard thermodynamic expression:
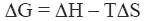
|
where ΔH and ΔS are the enthalpy and entropy changes respectively. In this chapter we will follow the common practice of using the native state as the reference state. Since both, the enthalpy and entropy changes are not constants but increasing functions of temperature, the Gibbs energy of stabilization of a protein needs to be written as:
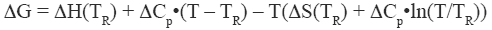
|
where TR is a convenient reference temperature, ΔCp is the heat capacity change, and ΔH(TR) and ΔS(TR) are the enthalpy and entropy values at that temperature. Because 60°C (333.15 K) is close to the median denaturation temperature of proteins, it has been chosen as a convenient reference temperature for comparing several proteins since that reference temperature minimizes extrapolation errors.
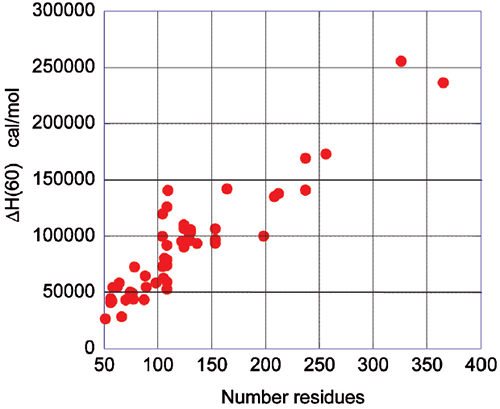
The energetics associated with the folding/unfolding of globular proteins exhibits great regularity, reflecting the common nature of the forces that stabilize the native structure. When the enthalpy change for different globular proteins is compared at 60°C, a clear correlation is seen between the enthalpy change and the size of the protein (number of residues). This phenomenon is illustrated in Figure 2 and provides an important benchmark for the interpretation of thermodynamic data for novel proteins. For example, a smaller enthalpy change than the one expected for a given size is an indication that the protein might have unstructured regions.
|
Upon protein unfolding, the major contribution to the enthalpy change arises from the disruption of intramolecular interactions (van der Waals, hydrogen bonds, etc.) and the parallel solvation of the interacting groups. On average, those contributions are similar for different globular proteins and therefore scale in terms of size. Minor contributions, like those associated with protonation/deprotonation processes are not sufficiently large to account for large deviations in the magnitude of the enthalpy or entropy changes. The change in heat capacity upon unfolding is also correlated with the size of the protein as seen in Figure 3.
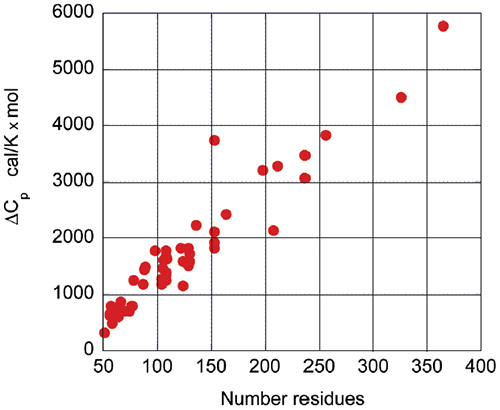
|
The heat capacity change is mainly the result of changes in hydration of groups that are buried from the solvent in the native state and become solvent exposed upon unfolding. It has been known for many years that changes in heat capacity associated with hydration are local in nature and scale in terms of the changes in the surface area accessible to the solvent.9-12 For globular proteins, the surface area that becomes buried in the native state is proportional to the number of residues in the protein. Thus, the enthalpy and the heat capacity changes can be used to evaluate the level of structure in a protein.
One of the most notable cases reported in the literature corresponds to the HIV-1 envelope glycoprotein gp120 (Ref13 and unpublished work from this laboratory).
gp120 is a protein of about 500 amino acids that undergoes a two-state unfolding with a Tm of 59.2°C, a value close to that expected for an average globular protein. The enthalpy change associated with the transition is 180 kcal/mol, which translates to a value of only 2.3 cal/g. This is less than half the expected value for the average globular protein at the same temperature, and corresponds to the unfolding of a protein of about 300 residues. The Tm value and the cooperative two-state transition suggest that gp120 has a well-structured core and regions of very low structure that do not contribute to the enthalpy of the unfolding process.
Before a molecular interpretation of the thermodynamic parameters can be made, it is necessary to assess if the protein under study has the required quality for analysis. Here, DSC also plays a critical role. A well-characterized protein batch may serve as reference for evaluating the quality of future batches. High quality recombinant proteins need to be not only free of impurities but also free of misfolded, denatured or partially denatured forms of the same protein. Conventional analytical techniques like gel electrophoresis, light scattering or mass spectrometry will not detect or quantitate misfolded protein forms in solution. For enzymes, specific acitivity measurements or active site titrations can evaluate the population of inactive forms, however for proteins that lack enzymatic activity this is a difficult task. Usually, the protein needs to be characterized simultaneously by several techniques including chromatography, electrophoresis, mass spectrometry, NMR, DSC, etc. Once a high quality, well characterized protein is obtained, it can serve as a standard. DSC provides an effective and rapid way to accurately evaluate a protein batch by comparison with the standard.
The DSC of a well characterized protein batch (the standard) will be characterized by three different parameters: 1) The Tm or location of the peak maximum; 2) The enthalpy or area under the curve; and, 3) The shape of the curve. For a new protein batch to be of the same quality, it should match the standard in those three parameters. The Tm is sensitive to the presence of ligands or cofactors and, therefore, a Tm shift, up or down, may be indicative of the presence or absence of ligands or cofactors. The area under the curve is the best indicator of the presence or absence of denatured forms of the protein. A smaller area is a clear indication that not all the protein is correctly folded. In fact, the fraction of protein that is correctly folded can be estimated from the ratio of the areas measured for the new protein and the standard. The shape of the curve reflects the folding/unfolding mechanism, and consequently, the presence of partially folded forms that also undergo thermal denaturation will affect this shape. An example of DSC results obtained with different batches of gp120 is shown in Figure 4. The area under the curve for each batch is proportional to the fraction of gp120 that is correctly folded, and equal to the fraction that is able to bind CD4, as determined by ITC.

|
Once protein partners have been characterized individually, the nature and consequences of their interaction can be evaluated and interpreted. There are different scenarios that need to be considered and all of them have different thermodynamic signatures. For example, one possible scenario is that of two well structured proteins that associate without inducing any significant conformational change; a second scenario could be the association of two well structured proteins that bind and induce a conformational change; a third scenario could be a protein that binds to another protein with unstructured regions that become structured as a result of binding. All of these situations can be interpreted by combining DSC and ITC data; or, in more complex situations, by incorporating structural data and structurally parameterized thermodynamics.14-16
In the absence of conformational changes, the binding energetics of two proteins is dominated by their interacting surfaces. The magnitude and sign of the enthalpy change depends on the nature of the interactions between the two surfaces. Literature values for different proteins range from exothermic to endothermic enthalpies. Since the areas buried from solvent upon binding are predominantly hydrophobic, the binding of two proteins is coupled to a negative change in heat capacity. The burial of a hydrophobic surface is also coupled to a positive entropy change, which in the absence of conformational structuring yield a net favorable entropy change.
The situation is very different if binding induces significant protein structuring. In this case, the binding thermodynamics is not dominated by the interacting surfaces that can be observed by x-ray crystallography. We will illustrate this case using again the binding of the HIV-1 envelope glycoprotein gp120 to its cell surface receptor CD4. This is the first event in HIV-1 infection and the interaction is a major target for drug development. As mentioned above, gp120 is a largely unstructured protein in its unliganded state, however the crystal structure only exists for gp120 in complex with CD4 and the Fab fragment of either of the monoclonal antibodies 17b or X5, where the N- and C-termini are truncated and most of variable loops are deleted.17,18
The first ITC studies, carried out by Myzska et al19 showed that binding of the soluble form of the CD4 receptor to gp120 is associated with an unusually large and favorable change in enthalpy that is opposed by a large unfavorable change in entropy. Subsequent studies in this laboratory provided a complete representation of the binding reaction.13 In this case, the largely unstructured gp120 molecule undergoes a massive structuring upon binding to CD4, which also gives rise to an unusually large and negative heat capacity change, reflecting the large number of amino acids buried from water.
The binding of a protein to another often induces conformational changes that initiate signaling cascades. In the case of gp120, the structuring induced by the binding of CD4 triggers the organization and concomitant competence of the chemokine co-receptor site in gp120. The organization of the binding site by CD4 eliminates the energy penalty for the co-receptor (CCR5 or CXCR4) resulting in a much higher binding affinity. The binding of CD4 to gp120 triggers the binding to the co-receptor and the subsequent chain of events that lead to the fusion of the viral and cell membranes. The increased affinity for the co-receptor site induced by CD4 has been thoroughly studied by ITC in this laboratory using the monoclonal antibody 17b, which binds to an epitope that overlaps the binding site for the chemokine receptor, CCR5.13,20 MAb 17b binds to gp120 with an affinity of 5nM in a process that is characterized by a large favorable enthalpy change (ΔH= -28.9 kcal/mol) that is opposed by a large change in unfavorable entropy (ΔS= -59 cal/(K• l mol)). The binding of CD4 to gp120 increases the affinity for 17b, which binds to the gp120-CD4 complex with sub-nanomolar affinity. The increase in affinity is associated with a smaller value for the change in enthalpy (ΔH= -10.1 kcal/mol) and slightly favorable entropy change (ΔS= 6.7 cal/(K • mol)). The smaller enthalpy change reflects the absence of a major structuring (it is already done by the prior binding of CD4) and the favorable entropy reflects the fact that the desolvation is no longer coupled to a large loss of conformational degrees of freedom. Figure 5 below shows the binding of 17b to gp120 alone and in complex with CD4. The change in affinity for the co-receptor (mimicked by 17b) can be quantitatively explained by the thermodynamics of CD4 binding to gp120. CD4 binds to gp120 with changes in enthalpy and heat capacity of -34.6 kcal/mol and -1.8 kcal/(K • mol), respectively; values with a magnitude reminiscent of protein folding rather than of binding reactions (Figures 2 and 3). In fact, by using the value for heat capacity change together with the unfavorable entropy change of -79 cal/(K • mol), it is possible to estimate that close to 130 residues in gp120 become structured upon CD4 binding (see Ref14).
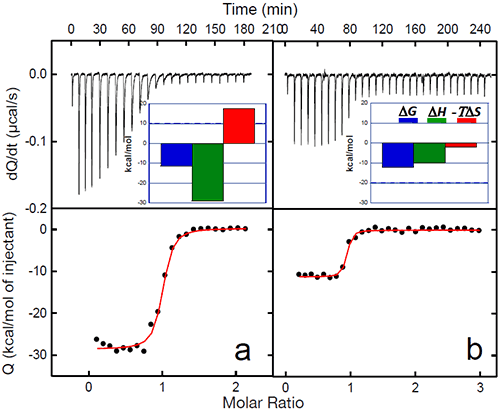
|
The co-receptor site in gp120 is largely unstructured in the unliganded protein and the direct binding of 17b is hampered by the required structuring. The co-receptor site in gp120 becomes structured and binding competent upon CD4 binding, thus removing the structuring requirement for the co-receptor and consequently, a much higher binding affinity.
Competitive inhibitors bind to the same binding site as the ligand protein, preventing it from binding. There are advantages and disadvantages in the development of this type of inhibitors. The main advantage is that the structure of the binding site is usually known, facilitating structure-based design. There are two main disadvantages: 1) the requirement for extremely high affinity; and, 2) the likelihood that the inhibitor acts as a surrogate ligand. Since protein/protein interactions often display high affinity, their disruption requires a very high affinity inhibitor, something difficult to achieve with a small molecule. Also, by binding to the same place as the original ligand, the possibility always exists that the inhibitor ends up eliciting the same effect as the protein ligand. A clear illustration of this situation has been reported for some competitive inhibitors of the CD4/gp120 interaction.
NBD-556 is a recently identified low-molecular weight compound21 that binds to gp120 and blocks the interaction with CD420. NBD-556 binds to gp120 with an affinity of 3.7 μM, i.e. about three orders of magnitude weaker than CD4. Despite its low affinity, the direct binding of NBD-556 to gp120 is associated with large favorable enthalpy and unfavorable entropy changes and a large negative change in heat capacity, a thermodynamic signature similar to that observed for CD4 binding. The ability of NBD-556 to compete with CD4 was studied in experiments where gp120 was titrated with CD4 in the absence and presence of NBD-556. The expected apparent affinity in the presence of a competitive inhibitor can easily be calculated using the following equation:

|
where Ka is the association constant for sCD4 in the absence of inhibitor, Ka, app is the expected apparent association constant for sCD4 in the presence of inhibitor, Ka,I is the association constant of the inhibitor and [I] is the concentration of inhibitor. The expected enthalpy change in presence of inhibitor, ΔHapp, is calculated according to:

|
where Fb is the degree of saturation of gp120 with the pre-bound inhibitor and ΔH and ΔHI are the enthalpy changes for the binding of sCD4 and the inhibitor, respectively.
The experimental values obtained from ITC experiments of sCD4 in the presence of NBD-556 could be well accounted for by equations 3 and 4, indicating that NBD-556 and CD4 compete for the same site in gp120. As in the case of CD4, binding of NBD-556 to gp120 structures the co-receptor site, enhances the binding affinity for MAb 17b and acts like a CD4 surrogate in cell-based assays. In fact, in CD4 negative cells, the presence of NBD-556 enhances HIV-1 infection by three orders of magnitude.20 This result clearly illustrates the danger of a competitive inhibitor that emulates the natural ligand in all its aspects.
Allosteric inhibitors do not compete with the protein ligand; rather, they inhibit the propagation of the signaling cascade by binding to a site along the allosteric pathway. As with competitive inhibitors, allosteric inhibitors also have advantages and disadvantages. The main advantage is that they don’t have to displace a high affinity protein ligand, and therefore their affinity requirement is not a stringent. The main disadvantage is that very often the allosteric site is unknown or not immediately obvious.
BMS-378806 (BMS-806) and its analogs22-24 are examples of low-molecular weight gp120 antagonists that inhibit infection with HIV-1 by an allosteric mechanism.
BMS-806 is potent and specifically inhibits infection with HIV-1 with an IC50 of less than 10 nM for most strains of HIV-1.
Because BMS-806 was selected in a high-throughput infectivity screen, its exact mechanism of entry inhibition has been controversial. BMS-806 was first believed to act primarily by blocking the gp120-CD4 interaction23,24, however, both calorimetric studies and viral inhibition assays carried out in concert point toward a different mode of action.20,25 The binding affinity of BMS-806 for gp120 is 43 nM and the process is mainly entropically driven (ΔS= 30 cal/(K • mol)) with an enthalpy change that is close to zero. These values are not atypical for the binding of small molecular weight inhibitors. The change in heat capacity associated with the binding of BMS-806 to gp120 is -292 cal/(K • mol), a typical value for the burial of a small hydrophobic molecule. Results from ITC experiments where gp120 was titrated with CD4 in absence and presence of BMS-806 deviated drastically from the expected values assuming competitive inhibition (equations 3 and 4). The affinity of CD4 for gp120 saturated with BMS-806 only decreased by a factor of 10 whereas a factor of 7000 would have been expected according to equation 3 if BMS-806 and CD4 competed for the same site in gp120. The ITC results agree with viral inhibition assays which confirm that BMS-806 also inhibits HIV-1 infection in CD4 negative cells.
The example of the HIV-1 glycoprotein gp120 and the efforts to develop HIV-1 entry inhibitors clearly illustrates the issues associated with the development of small molecular weight inhibitors of protein/protein interactions.
An inhibitor that competitively blocks the CD4-gp120 interaction needs to have an extremely high affinity in order to displace CD4 that has an affinity of about 10 nM. Furthermore, competitive displacement of CD4 also requires that the ligand binds without triggering the conformational changes that ultimately lead to enhanced co-receptor binding and viral entry. NBD-556 inhibits CD4 binding to gp120 competitively but, because of its low affinity, its effect on CD4 binding is only observed at very high concentrations. NBD-556 also acts like a CD4 mimetic, triggering the same conformational changes in gp120 that lead to co-receptor activation and, consequently, very serious unwanted effects.
The affinity of BMS-806 for gp120 is slightly lower than that of CD4, however, BMS-806 is an allosteric inhibitor and does not need to compete with CD4 to achieve potent inhibition of viral entry.
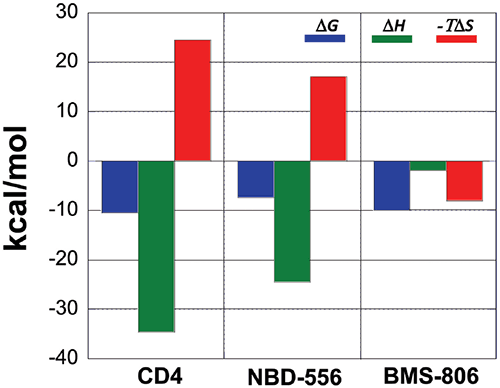
A comparison between the binding thermodynamics for different inhibitors can serve as a guide in the selection and optimization of inhibitors of protein-protein interactions. Figure 6 shows the thermodynamic signatures for the binding of CD4, NBD-556 and BMS-806 to gp120. Because the binding of CD4 to gp120 is associated with a large structuring of gp120, its thermodynamic signature is very characteristic and composed of a large negative enthalpy change that is opposed by a large unfavorable entropy term. Any inhibitor that competes with CD4 and triggers a similar conformational change will also activate the co-receptor site and exhibit a thermodynamic signature similar to that observed for CD4. NBD-556 is an example of such an inhibitor.
An inhibitor that does not induce the structuring of gp120 will not exhibit that thermodynamic signature. For example, the allosteric inhibitor BMS-806 that binds to a different site without inducing any conformational changes can easily be distinguished because of its completely different thermodynamic signature. Also an inhibitor that competes with CD4 without inducing a structuring in gp120 will have a thermodynamic signature approaching that of BMS-806, i.e. a thermodynamic signature characterized by a smaller or even favorable entropy change. In this respect, we have been able to determine that sites that can be used to enhance binding potency do not necessarily overlap with sites that initiate allosteric effects.
Distinguishing between those sites, using algorithms like CORE_BIND26 or similar, should allow the development of competitive inhibitors that do not act as surrogate ligands.
|
This work was supported by grants from the National Institutes of Health (GM56550 and GM57144) and the National Science Foundation (MCB0641252).
