Size-exclusion chromatography (SEC) or gel permeation chromatography (GPC) is frequently used in bioscience laboratories to characterize purified and recombinant proteins. This technique is used to measure molecular weight or to resolve oligomer mixtures. This is been done by creating a calibration curve of standard globular proteins and comparing the elution time of an unknown to the standards to derive its molecular weight. This traditional method is limited in that it is based on a stable relationship between molecular weight, size and shape or hydrodynamic volume. However this relationship varies significantly between proteins introducing unquantifiable inaccuracies in all measurements made in this way.
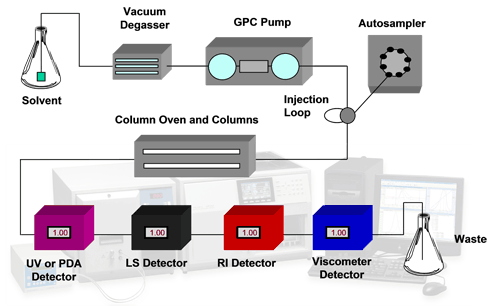
Modern analytical systems are capable of more than sample detection with a single concentration detector. The use of multiple detectors including refractive index (RI), ultraviolet (UV), light scattering (LS) and viscometry (IV) allows extensive characterization of protein samples in a more absolute way than was previously possible. RI and UV both allow accurate concentration measurements to be made. A combination of these allows conjugation analyses to be performed. Light scattering is used for the measurement of molecular weight and viscometry measures the intrinsic viscosity of a sample which is an indicator of structural changes. A combination of these also allows molecular size to be calculated. This is called tetra detection. A typical tetra detector analytical SEC system is shown in the schematic in figure 1.
|
This application note describes some of the most common protein applications for which an advanced analytical SEC system is used.
Individual proteins have a very well defined molecular weight. Once synthesized within a cell, and following any post-processing, such as cleavage or glycosylation, a protein's molecular weight will be fixed with minimal variation.
Measuring the molecular weight of a novel protein is interesting of itself. Different proteins can have significantly different molecular weights, so measuring the molecular weight of a purified protein is an excellent way of confirming that the purified sample contains the protein of interest. During production of a recombinant protein, confirming the molecular weight to be the expected value is a good indicator that it is being produced correctly by the host cell line.
Measuring molecular weight is therefore of interest both academically and practically as an indicator of product or sample quality.
Figure 2 shows an example of alcohol dehydrogenase, a typical standard protein. Its molecular weight has been measured as 149 kDa and is constant across the eluted peak. A very small secondary peak can also be observed and its molecular weight has been measured to be slightly over 300 kDa. The shape of the molecular weight trace, with a small plateau but increasing at the leading edge, suggests that this peak is a dimer and the sample contains larger aggregates.

|
The activity of any protein is defined by the final structure or conformation that it takes. Quaternary structure defines the final complex formed when multiple protein chains come together to form a single unit. This too can be assessed by analytical SEC if the molecular weights of the individual protein are known or measured. Proteins are individually monodisperse meaning their molecular weights are stable across a peak as they elute from a SEC column. As multiple protein molecules come together to form oligomers, the measured molecular weight will increase discretely resulting in step changes of molecular weight within the chromatogram.
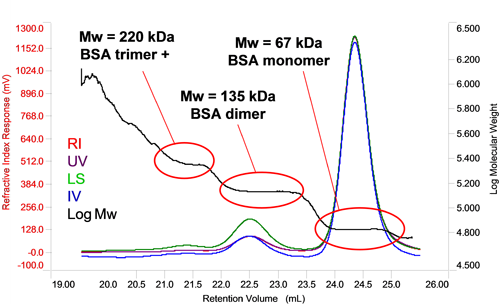
In the case of homo-oligomers, when a number of identical proteins come together, the measured molecular weight will be a multiple of the monomer molecular weight allowing the oligomer number to be determined via a simple calculation. In figure 3 it can be seen that the molecular weight of BSA increases in steps between each peak. Each of the molecular weights corresponds to a multiple of the monomer molecular weight, in this case, two and three fold indicating the presence of dimers and trimers. In the case of the trimer, the slightly higher than expected molecular weight and that fact that it is increasing at earlier retention volumes within the peak suggests this peak also contain some larger oligomers. The proportion of each can, of course, be individually measured by the RI or UV detector to determine the overall composition of the oligomer mixture.
|
Protein aggregation is a common result of sample treatment such as prolonged storage or freeze/thaw cycles. The structure of a protein is determined by Van der Waals forces, hydrogen bonds, disulphide linkages and hydrophobic interactions. Changing the buffer conditions can disrupt the delicate balance that holds the structure together to reveal buried regions of the polypeptide chain. These regions can interact with those on other proteins to form larger complexes of misfolded proteins. Hydrophobic regions are especially susceptible to this effect. Aggregates are often strongly held together by these forces meaning that their formation is irreversible and they promote further aggregation. The activity of the proteins will be lost and they may reach the point when they are no longer soluble.
Protein aggregates tend to have a very high molecular weight and are often very polydisperse as the complexes formed are very unlikely to become stable structures of predictable sizes.
In SEC separations, aggregate peaks can sometimes appear in the void volume of the column as they are too large to penetrate any of the pores in the packing material. Their molecular weight may have limited meaning given their high polydispersity but it can, nevertheless, be measured along with their concentration using refractive index or UV.
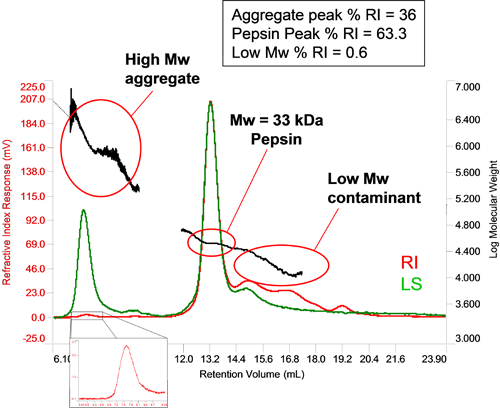
The relative sensitivity of light scattering and concentration detectors means that in the case of very small amounts of very high molecular weight material, the concentration may be below the limit of the concentration detector, in which case the light scattering effectively acts as a 'first responder' to the formation of aggregates. If their concentration is high enough, then their concentration can be measured. Figure 4 shows a sample of pepsin with a small amount of aggregated material. Its large molecular weight and high polydispersity are typical of aggregated proteins. The small RI peak allows the concentration to be determined as a proportion of the total material and is seen to make up 0.6% of the sample. In this particular sample, some low molecular weight material has also been identified and composes 36% of the sample.
|
Intrinsic viscosity is inversely proportional to molecular density (or partial specific volume). This can be measured using a viscometer. A combination of this and the molecular weight data from light scattering also allow the molecular size to be determined. These two values are very useful for assessing structural changes between samples. A change in size and intrinsic viscosity with a constant molecular weight is a clear indication of a change in structure and from an increase or decrease in density, an idea of the structural change can be reached.
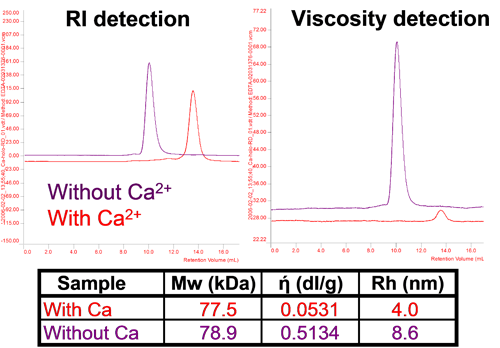
For proteins, structure determines function, and changes in or loss of structure result in changes in or loss of activity. Proteins can change their conformation in different conditions and this can be assessed using this technique. Figure 5 shows adenylate cyclase toxin which changes its size and intrinsic viscosity (IV or ή) significantly without a change in molecular weight with changes in calcium concentration. In the absence of calcium ions, the protein is unfolded with high IV and a larger size. With addition of calcium, the protein assumes its native conformation with a smaller size and a considerably lower IV. It is worth noting that the change in IV is proportionally much larger than the change in size showing its higher sensitivity to these changes.
|
Analytical SEC has undergone significant development since its inception. While measurements of molecular weight were the original goal of the technique, these can now be made in an absolute sense, without relying on column calibration. Furthermore, concurrent additional measurements of concentration, intrinsic viscosity, size and conjugation provide extra information that make multi-detection SEC an invaluable tool for bioscience laboratories.
All of this work was performed on a Viscotek TDAmax system.